Metabolomics Data Analysis Service — From Raw Spectra to Biological Insight
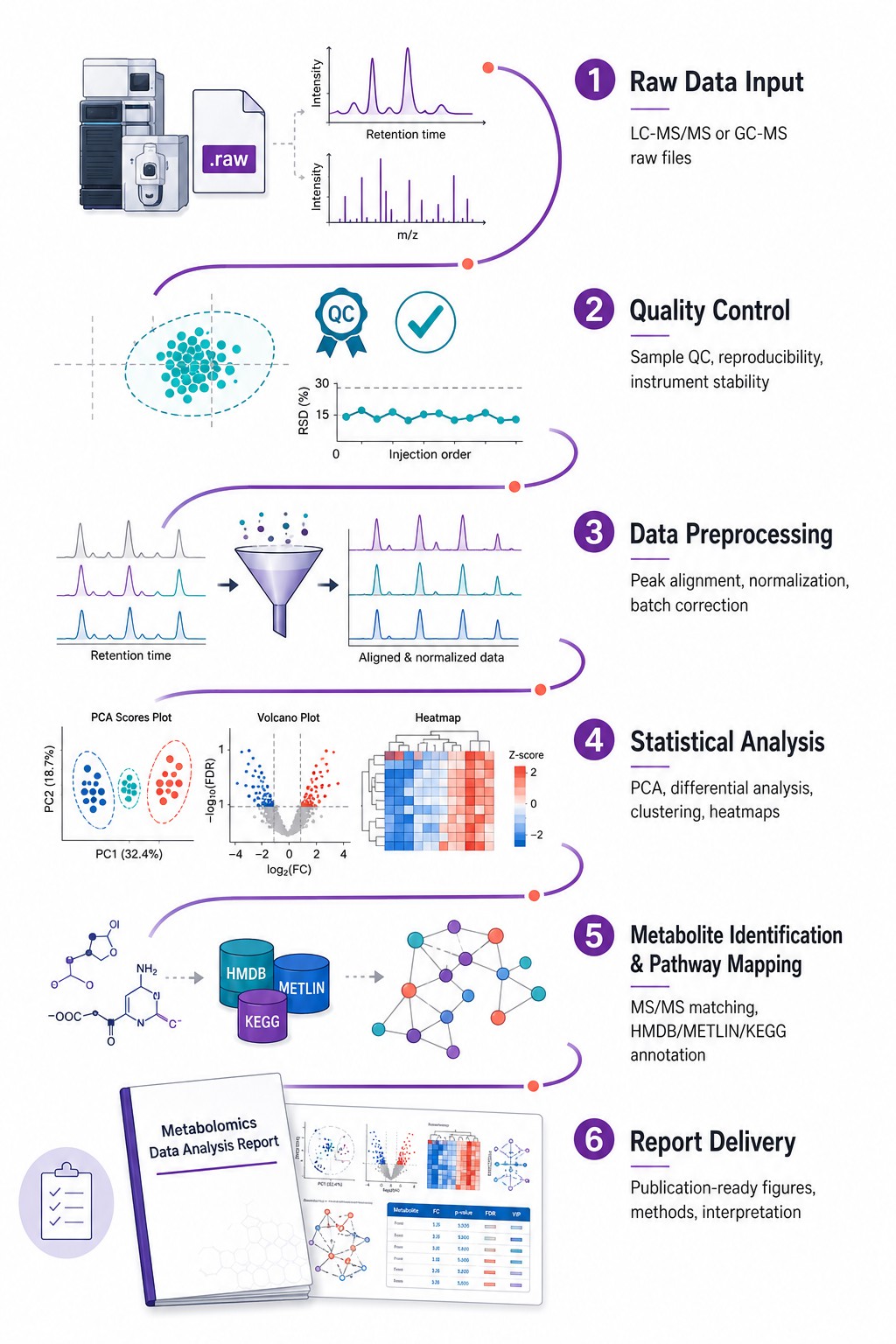
You have the data. We turn it into answers. Our bioinformatics team handles every computational step — preprocessing, statistics, metabolite identification, pathway enrichment, multi-omics integration — and delivers publication-ready figures, methods documentation, and a biological interpretation you can defend to reviewers. Accept raw instrument files from any vendor, processed peak tables, or public repository datasets — platform-agnostic, fully reproducible, all parameters documented.
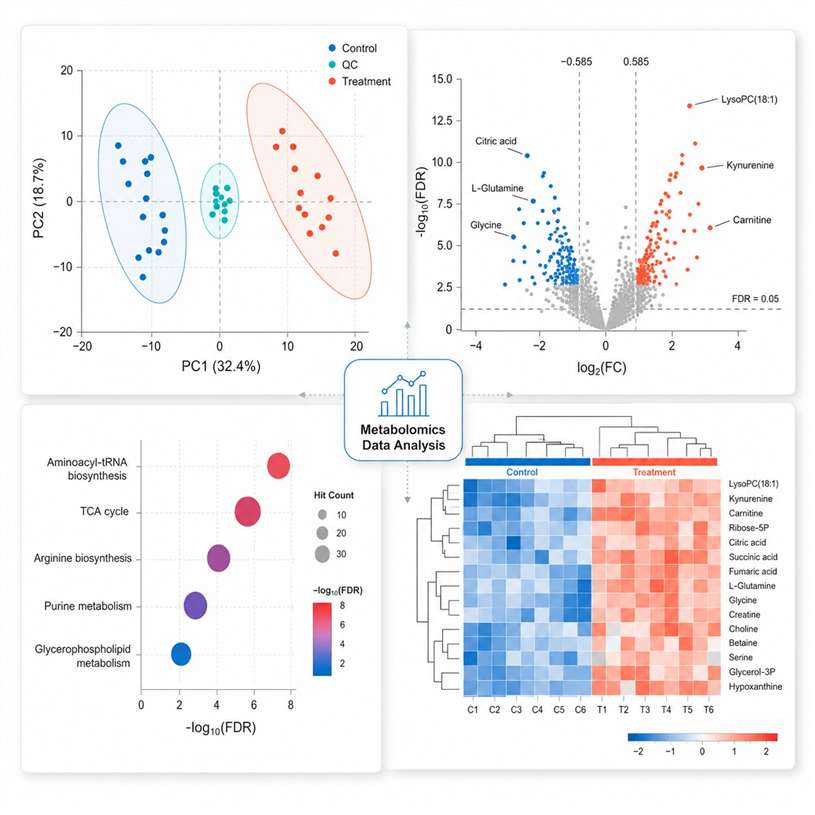
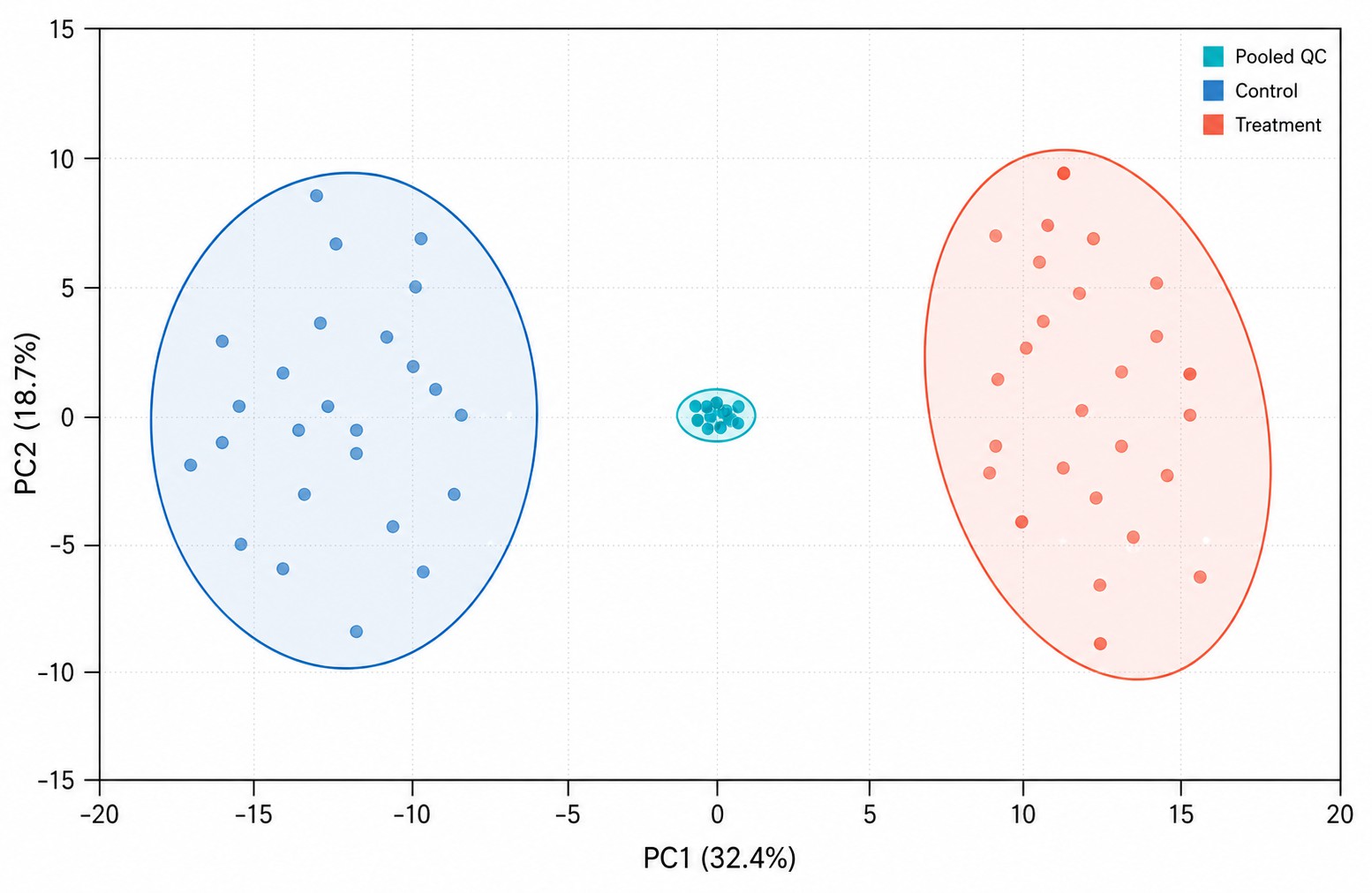
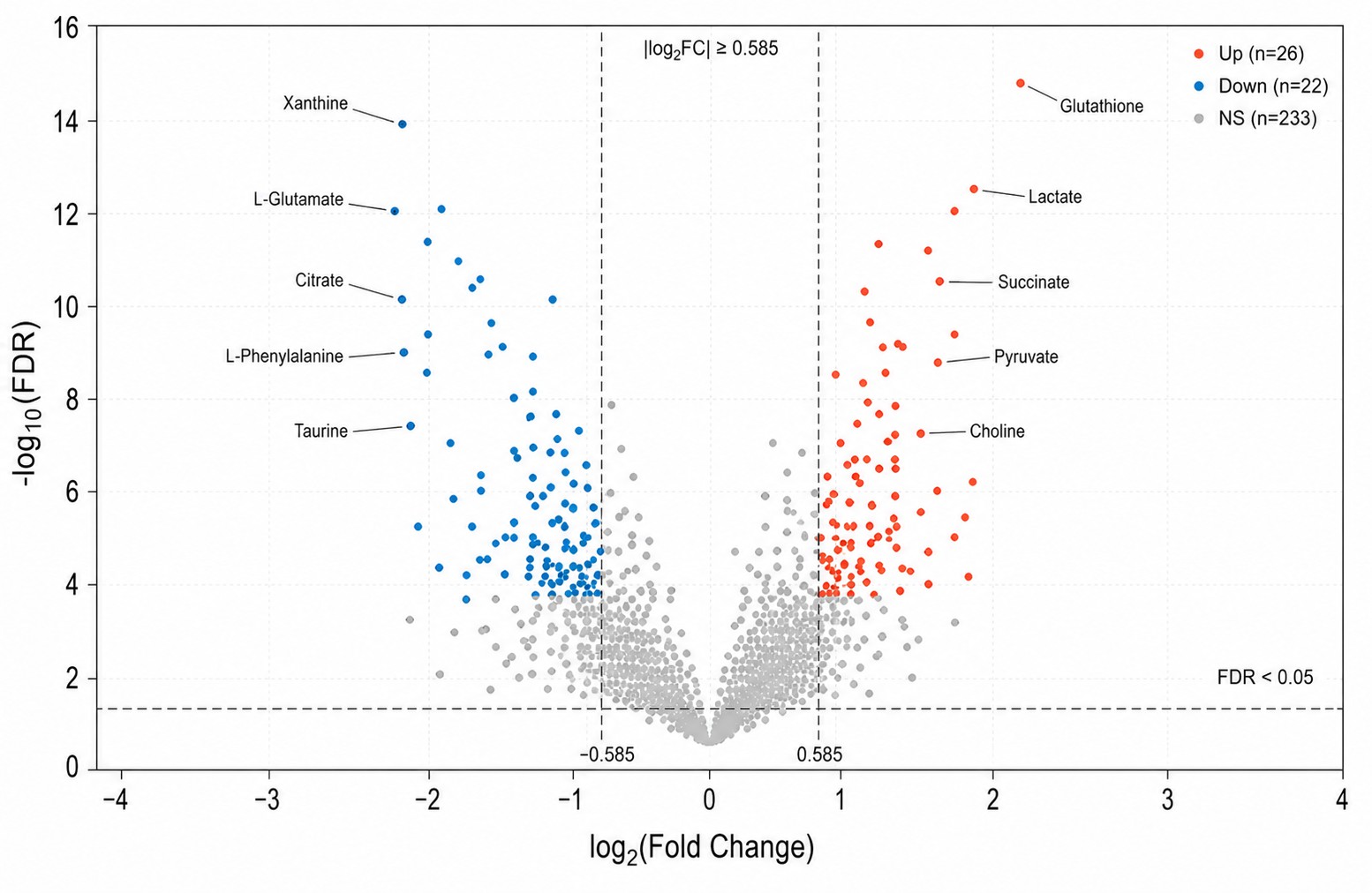
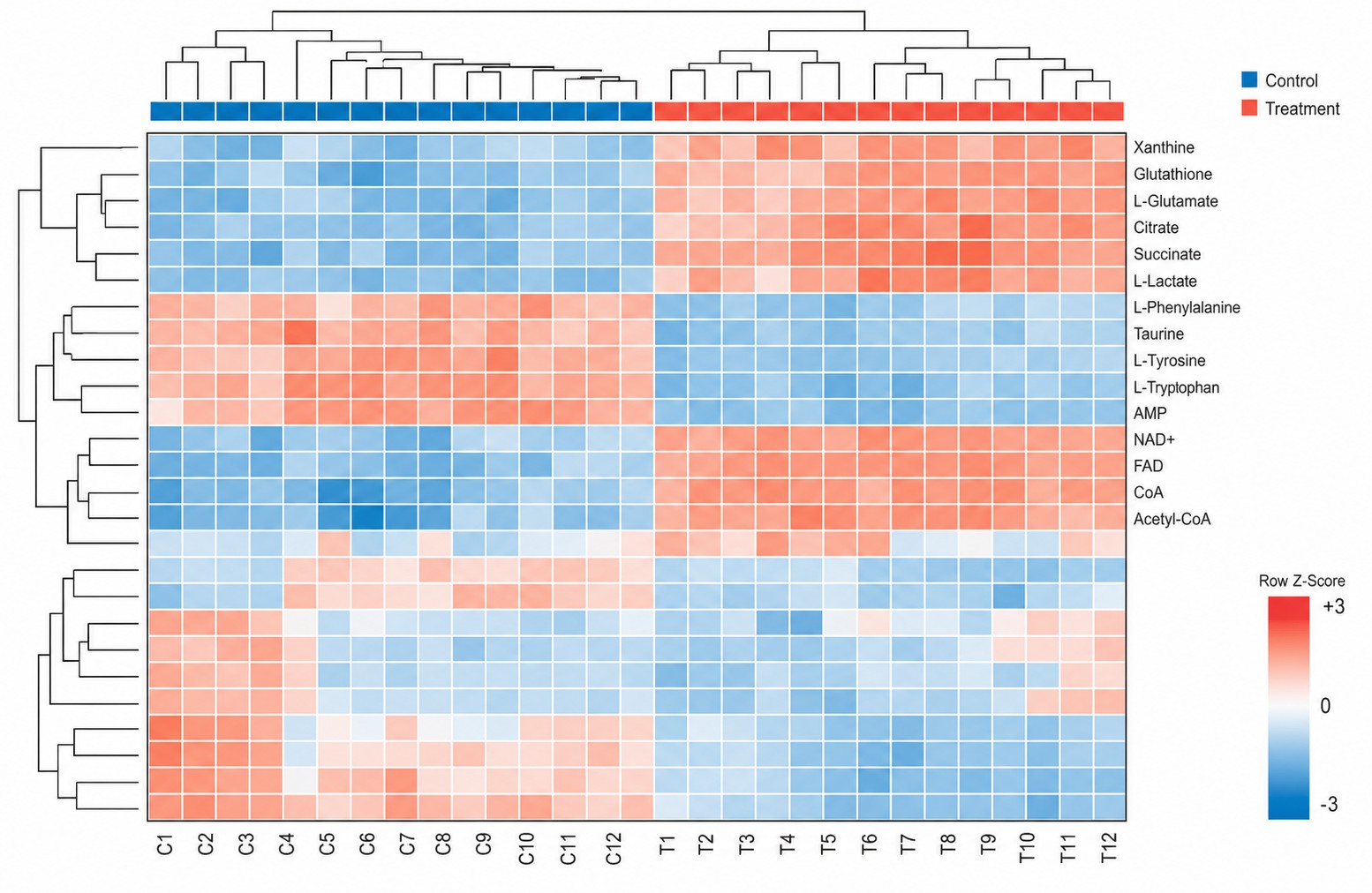
Multivariate statistics: PCA, PLS-DA, OPLS-DA with permutation testing (n≥1,000), hierarchical clustering, volcano plots
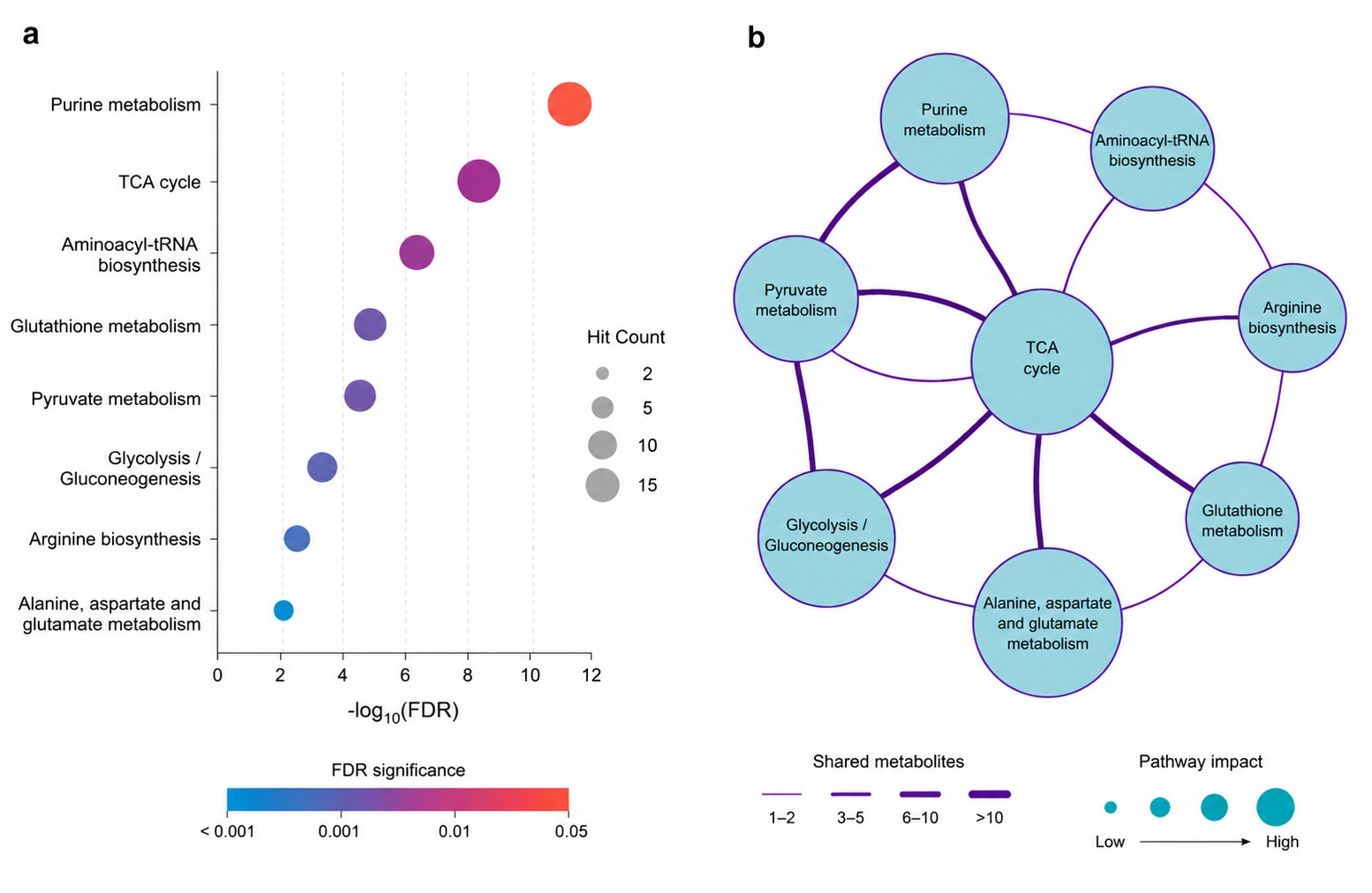
Pathway enrichment (KEGG, Reactome) and metabolic network analysis with FDR correction at every step
Multi-omics integration: DIABLO, MOFA+, O2PLS — metabolomics + proteomics + transcriptomics + lipidomics
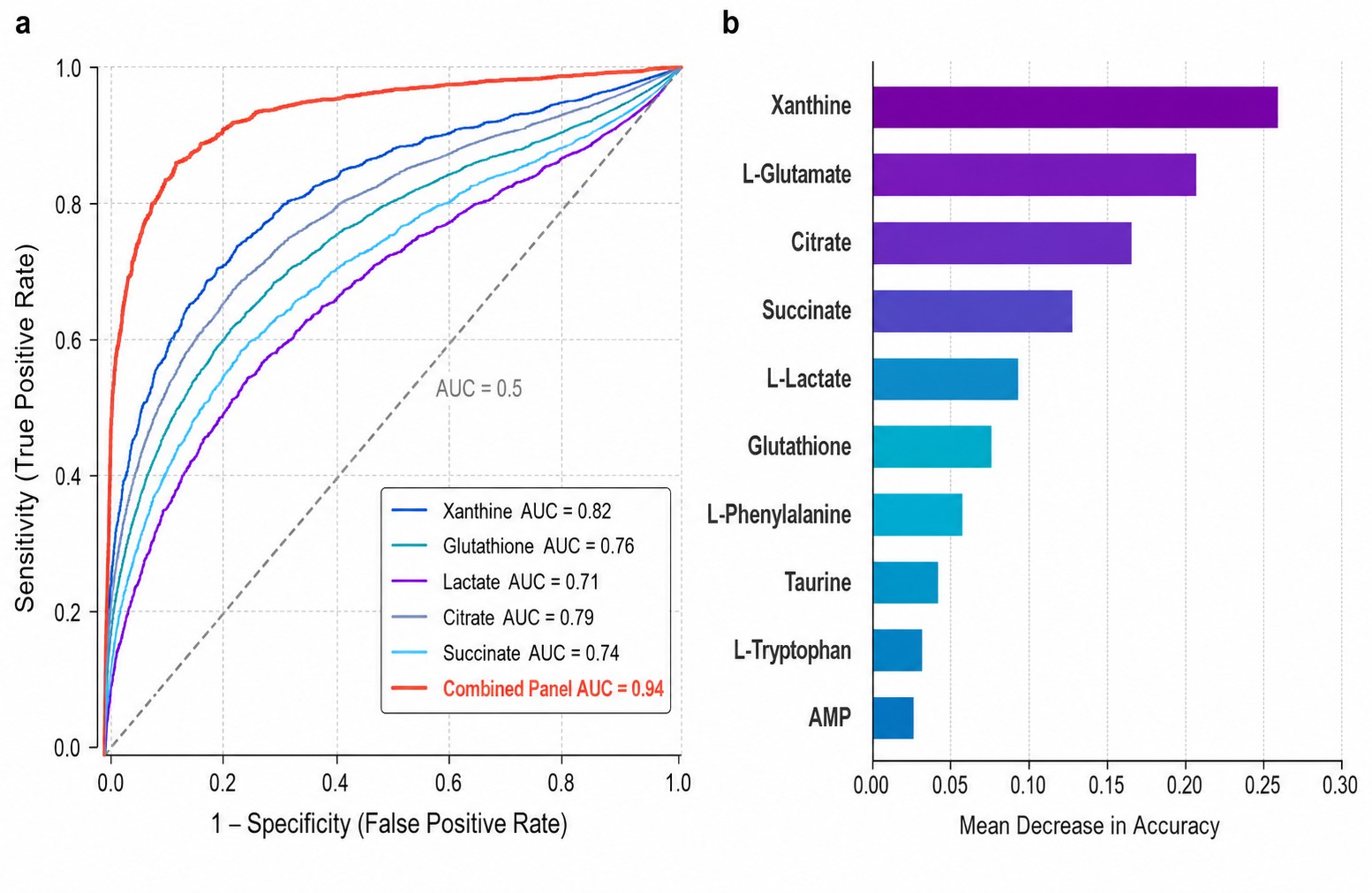
Machine learning & biomarker discovery: random forest, SVM, XGBoost, ROC analysis, multi-metabolite panels
Platform-agnostic: .mzML, .raw, .d, .wiff, .cdf, .csv — your data or ours, same rigorous pipeline